중국 의약품 등록 신청 접수 방법
#중국 의약품 등록 신청 접수 방법
중국에서 의약품을 허가받고 팔기 위해선 상당히 복잡한 절차와 긴 시간이
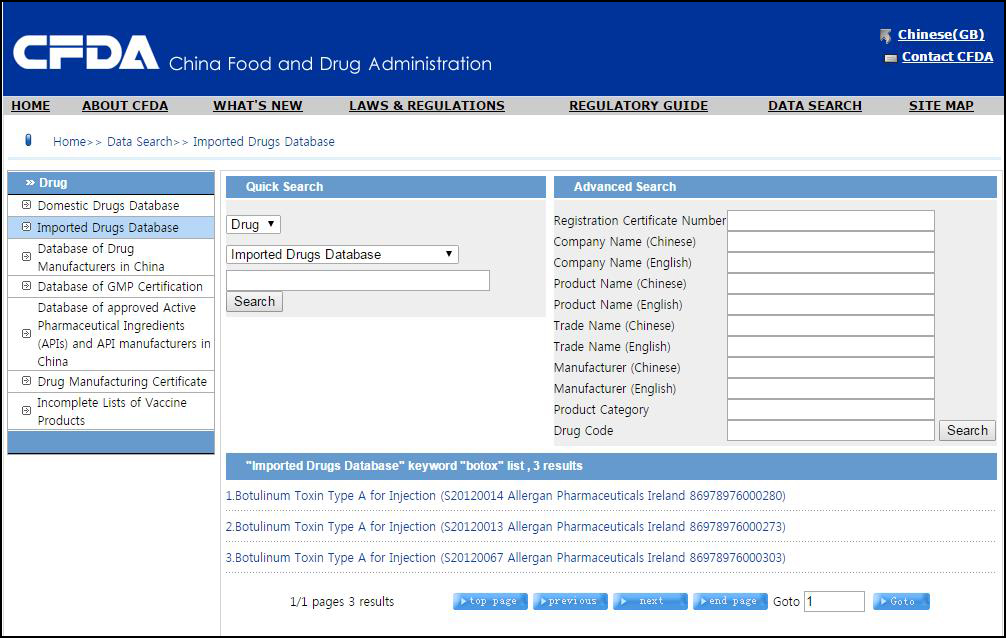
필요합니다. 제가 상하이에서 근무할 때도 미국의 엘러간사의 보톡스와 쥬비덤,
한국의 경우 필러인 이브아르 정보만 정식 허가를 받아 판매되고 있었으니까요.
그동안 미디어에서 우리나라 보톡스와 필러의 중국 수출 및 판매량이 엄청나다고
국내에서 대대적으로 홍보를 하곤 했는데 대부분이 불법적인 루트로
유통된 물품입니다.
- 중국 CFDA 서면 접수
国家药品监督管理局药品审评中心
关于CDE 办事指南 主任信箱 机构职能 关于审评 在线办理
www.cde.org.cn
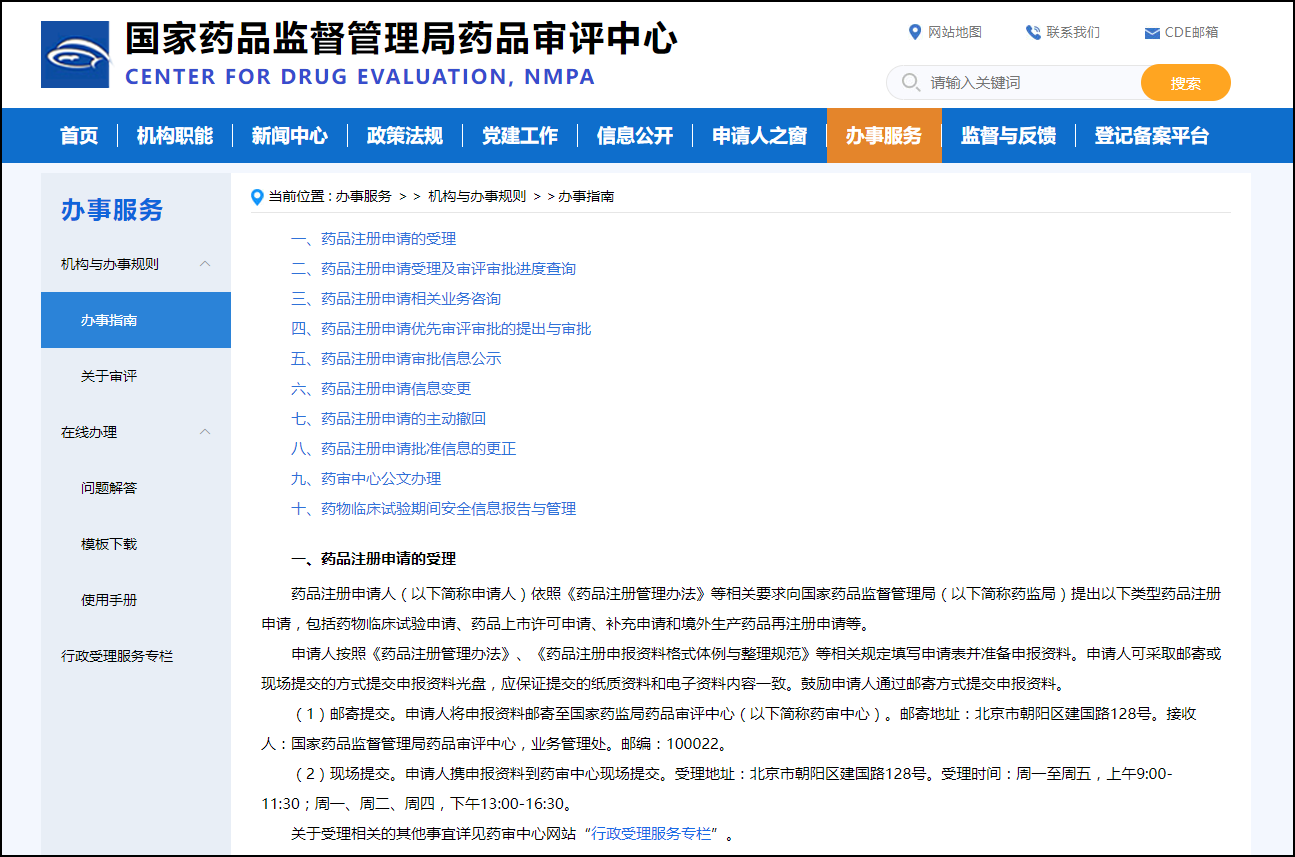
가장 먼저 정식 의약품 허가를 받기 위해선 중국 CFDA에 서면 접수를 해야 합니다.
서면 접수 요령은 위에 제가 링크해 드린 사이트를 방문하시어 진행을 해주시면
됩니다.
오늘 소개해 드릴 모든 사이트들이 예전엔 영어 버전을 제공하였으나 이제 허가를 더
빡빡하게 하기 위해선지 아니면 막기 위해선지 몰라도 전부 폐쇄되었습니다.
중국 허가를 위해선 중국 직원 고용이 필수가 되었습니다.
#중국 의약품 등록 신청 접수 방법 10단계
1. 의약품 등록 신청 접수
약품 등록 신청인(이하 신청인이라고 함)은 "약품 등록 관리 조치"의 관련 요구 사항에 따라
국가 약품 관리국(이하 약품 관리국)에 다음 유형의 약품 등록 신청서를 제출해야 합니다.
의약품임상시험신청, 의약품품목허가신청, 해외의약품 추가신청 및 재등록 신청 등
신청인은 "의약품 등록 관리 조치" 및 "의약품 등록 신청 문서의 형식 및 조합 기준"의 관련 규정에
따라 신청서를 작성하고 신청 자료를 준비합니다.
지원자는 지원 자료의 CD-ROM을 우편 또는 현장제출로 제출할 수 있으며, 지원자는 제출된 종이자료의
내용이 전자자료의 내용과 일치하는지 확인하여야 한다.
지원자는 우편으로 지원 자료를 제출하는 것이 좋습니다.
(1) 우편 주소: 北京市朝阳区建国路128号
수취인: 国家药品监督管理局药品审评中心,业务管理处
우편번호: 100022.
(2) 현장 제출 주소 : 北京市朝阳区建国路128号
접수 시간: 월요일~금요일(오전 9시~11시 30분)
월요일, 화요일, 목요일(오후 13시~16시 30분)
2. 의약품 등록 신청 접수 및 심사, 승인 진행 조회
검토 과정의 투명성을 보장하기 위해 의약품 평가 센터는 수행 중인 작업의
진행 상황에 대한 질의를 제공합니다.
웹사이트의 정보 공개에 있는 '受理品种信息(허가 품목 정보)' 및 审评任务公示(승인과정)을
통해 의약품 등록 신청의 전반적인 승인 상태와 다양한 의약품 등록 분류 시퀀스의
검토 및 승인 진행 상황을 확인할 수 있습니다.
3. 의약품 등록신청 관련 업무상담
신청인은 「의약품 연구개발 및 기술심사 교류 및 교류에 관한 관리조치」(2020년 제48호)에
따라 의약품 등록 신청 수정, 연구개발, 심사승인에 관한 사항을 상담할 수 있다.
이는 다음과 같이 시행할 수 있습니다.
(1) 일반적인 문제에 대한 상담
접수, 연구개발, 심사 및 승인절차에 관한 일반적인 문의는 의약품평가원 홈페이지의
申请人之窗(신청인 창구)의 일반 문의를 통해 상담이 가능합니다.
기술 검토 프로세스의 주요 결정 과정을 포함합니다. 이 부분부터는 신청인이
발급받은 로그인 아이디와 비밀번호가 필요합니다.
기술적인 문제의 경우 CFDA에서 질의를 받은 후 영업일 기준 15일 이내에 피드백을
제공하고 비기술적인 문제의 경우 영업일 기준 10일 이내에 피드백을 제공합니다.
(2) 주요 기술 문제에 대한 협의
의약품 연구개발 과정에서 핵심기술사항에 대해서는 「의약품연구개발 교류 및 교류관리 방안」에
의거 의약품평가센터 홈페이지의 申请人之窗(신청인 창구)를 통해 질의할 수 있다
4. 우선 심사 의약품 승인 요청
'의약품 등록 관리를 위한 조치' 및 '획기적인 치료 약물 검토를 위한 작업 절차'(2020년 82호)를
포함하는 3개의 문서에 의한 국가 식품 약품 감독 관리국의 발표에 따라
임상적 가치가 분명한 약품에 경우 우선 심사를 신청할 수 있다.
승인 절차:
(1) 주요 감염병 및 희귀질환의 예방 및 치료를 위한 임상에 시급히 필요한 약품 및 개량신약
(2) 아동의 특성에 부합하는 아동용 의약품의 새로운 품목, 제형 및 규격
(3) 질병 예방 및 통제를 위해 시급히 필요한 백신
(4) 획기적인 약물 치료 프로그램에 포함된 약물
(5) 조건부 승인 대상 의약품
(6) 국가약품감독관리국이 규정한 우선심사 품목
품목 허가 신청서를 제출하기 전 품목 허가 신청인은 품목 허가 심사원과 소통하여,
대상 확인 후 품목 허가 신청서와 전자 우선 심사 승인 신청서를 제출하여야 한다.
신청자가 제출한 우선 심사 승인 신청에 대해 优先审评公示(우선심사 신청 리스트)를 통해
신청 내용을 확인 할 수 있고 승인된 품목은 5일 동안 의약품심사평가원
홈페이지 '拟优先审评品种公示(우선심사)' 란에 공지된다.
5. 의약품 등록신청 승인 정보 공고
의약품 심사 센터는 시판 허가 품목에 대한 정보를 홈페이지에 공개하고, 시판허가 후
6개월 이내 심사 보고서(신청인의 기술비밀에 관한 내용은 공개하지 않음) 및
의약품 사용설명서를 게재한다. 일반인은 의약품평가원 홈페이지 '信息公开(정보공개) '의
'上市药品信息(등재의약품 정보)'란을 통해 조회할 수 있다.
6. 의약품 등록 신청 정보의 변경
의약품 판매 허가 신청 심사 기간 동안 의약품의 안전성, 유효성 및 품질 관리 가능성에
영향을 미칠 수 있는 중대한 변경 사항이 있는 경우 신청자는 원래 등록 신청을 철회하고
추가 조사 후 다시 신청해야 합니다.
신청인의 이름 및 등록 주소의 변경이 기술 심사 내용과 관련되지 않은 경우
서면으로 약물 평가 센터에 통보하고 관련 증빙 자료를 제출해야 합니다.
7. 의약품 등록신청의 자진취소
약품 등록 신청 심사 승인 과정에서 신청인이 해당 등록 신청을 철회할 경우
신청인은 공식 문서로 등록 신청 접수부서에 철회신청서를 제출할 수 있다.
8. 의약품 등록 신청 승인 정보의 정정
신청인이 약품 등록 신청서의 승인 문서(첨부 파일 포함)의 내용이 정확하지 않다는 것을
발견한 경우 " 일부 약품 관리 승인 항목에 대한 승인 절차 조정에 관한 국가 약품 관리국의
결정"( 국가약품감독관리국 명령 제31호) 공문서 형식으로 등록신청 심사승인부서에
정정신청서를 제출한다.
9. 의약품 평가 센터 공문서 취급
신청자는 현장 또는 우편을 통해 약물 평가 센터에 공식 문서를 제출할 수 있으며
제출 주소는 위 1번을 참조하세요.
- 확인사항
(1) CFDA는 센터의 업무와 관련된 문서만 받습니다.
(2) 중복 제출로 인한 자원 낭비를 방지하기 위해 특별한 경우를 제외하고
다중 제출하지 않는다.
(3) 공문서는 분명한 목적과 유창한 언어, 하나의 사항에 대한 하나의 문서를 제출해야 한다.
(4) 해당 사항이 특정 의약품 등록 신청과 관련된 경우, 공문서에 등록 접수 번호, 제품명,
신청 항목 및 기타 정보를 기재한다.
(5) 공문서에 제목을 표시하고 문서의 끝부분에 관인을 날인하고 등재연월일을 표시한다.
사본이나 팩스는 받지 않습니다.
(6) 원활한 연락을 위해 공문에 담당자의 이름, 휴대폰 번호, 유선 전화번호 및
기타 연락처 정보를 반드시 기재하십시오.
10. 의약품 임상시험 중 안전성 정보 보고 및 관리
'의약품 등록 관리 조치' 및 '의약품 임상 시험(시험) 중 안전성 정보 평가 및 관리 기준'의
관련 요구 사항에 따라 신청인이 의약품의 임상 시험을 수행하도록
승인된 후 (중국 전통 의약품, 화학 약품 및 생물학적 제품 포함), 임상 시험을 수행해야 합니다.
시험 기간 동안 안전 위험 관리의 주요 책임은 의심되고 예기치 않은 심각한 부작용 (SUSAR) 및
기타 잠재적으로 심각한 부작용에 대해 신속한 보고서를 제출하는 것입니다.
(1) 빠른 보고서
신청자는 '의약품 임상 시험 중 안전성 데이터의 신속한 보고를 위한 표준 및 절차'의 요구 사항에
따라 지정된 시간제한 내에 임상 시험 중 SUSAR 개별 사례 보고서를 제출해야 합니다.
보고서는 약물감시 전자전송시스템(PV 시스템) 또는 의약품평가센터 홈페이지의
申请人之窗(신청인 창구) 란을 통해 제출해야 합니다.
(2) 연구개발 중 안전성 업데이트 보고
신청인은 '연구개발(시험) 중 안전성 업데이트 보고 관리기준'의 관련 요구사항에 따라
DSUR을 기한 내에 작성하여 제출하여야 하며, 申请人之窗(신청인 창구)란을
통해 보고서를 제출하여야 한다.
#중국 의약품 빠른 허가를 위한 팁
중국에서 의약품 허가를 받기는 하늘의 별따기입니다. 이런 복잡한 절차 이외에도
적절한 로비가(?) 따라가야 하기 때문이죠.
또한 최근 해외 의약품에 대한 허가도 상당히 빡빡합니다.
자신들이 카피약을 만들어서 대체하기 위해서죠.
그래서 허가를 낼 때 용도가 다양하다고 해서 이것저것 제출하기보다는 가장 허가받기
쉬운 항목으로 일단 허가를 받은 이후 용법을 늘려가는 것이 유리합니다.
톡신의 경우에도 원래 사시 및 근육 경련에 사용된 것이 본래의 목적이다 보니 주름
개선의 목적으로 허가를 받기보다 본래의 치료 목적으로 허가를 먼저 신청하는 것이
유리합니다.
물론 이렇게 허가 난 제품을 다른 용법으로 사용하다 걸리면 문제가 될 수도 있지만
허가받지 않은 약물을 사용하는 것이 더 큰 문제가 되므로 일단 다른 용법이라도 먼저
허가를 받게 되면 시장을 확장하는데 훨씬 유리합니다.
'병원마케팅' 카테고리의 다른 글
| 화장품 수출을 위해 준비해야 하는 '전성분표'란? (0) | 2023.06.04 |
|---|---|
| 중국 비자 멀티플 비자를 받기 위한 초청장 샘플 공유 (0) | 2023.05.16 |
| 중국 외자 의료 기구 진출 관련 기본 전략 (2) | 2023.05.16 |
| 페이스북 광고 집행하는 방법_기초편 (0) | 2023.04.28 |
| 로컬 병원(피부과, 성형외과) 콜센터용 기본 매뉴얼 Vol.3 (0) | 2023.03.13 |
